
When it comes to bringing new therapies to patients in the U.S., two key FDA pathways are often discussed: New Drug Applications (NDAs) and Biologics License Applications (BLAs). While both serve the same ultimate goal—getting safe and effective treatments to market—they follow very different paths. Understanding the differences between NDAs and BLAs is crucial for companies developing new therapeutics, particularly as the biotech landscape continues to evolve.
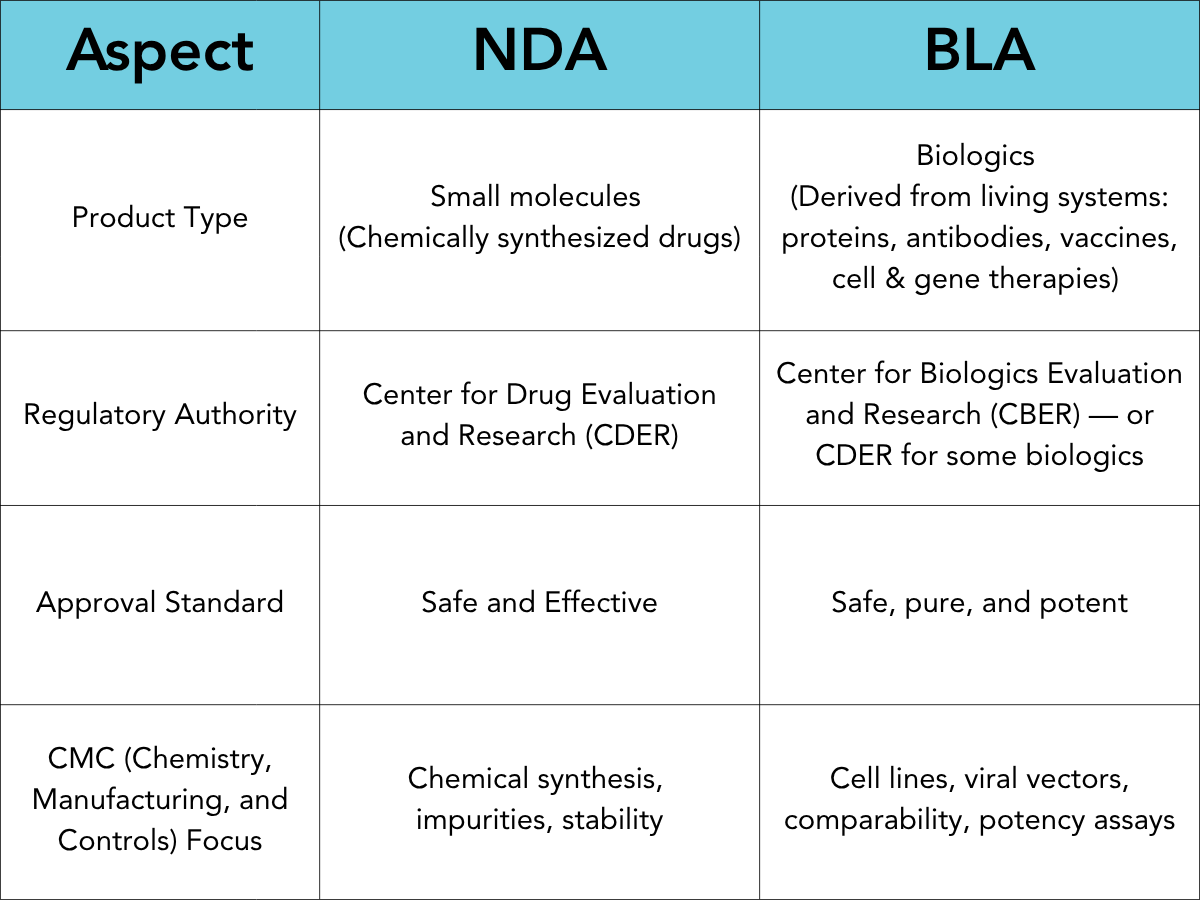
NDA vs. BLA: A Side-by-Side Comparison
Here’s a high-level overview of how these two regulatory pathways compare:
Why These Differences Matter
The distinction between NDAs and BLAs goes far beyond terminology—it has real implications for how a product is developed, manufactured, and ultimately reviewed by the FDA.
1. Development Strategy
Biologics often require more complex preclinical models and extended timelines to demonstrate potency and immunogenicity. These complexities influence early development decisions, clinical trial design, and regulatory interactions.
2. Manufacturing Controls
While small molecule drugs are chemically synthesized and relatively stable, biologics are produced in living systems, which introduces variability. Manufacturers must tightly control and document every aspect of the production process, from cell line development to lot-to-lot consistency.
3. Depth of Documentation
BLAs often require deeper characterization of the product, including details on comparability, bioactivity, and potency assays—especially for novel modalities like gene and cell therapies. These requirements increase the documentation burden and demand specialized expertise in biologics CMC.
The Bottom Line
As more companies pivot toward complex biologics, understanding the unique regulatory requirements of BLAs becomes essential. Success in this pathway depends not only on scientific innovation but also on a deep understanding of the regulatory landscape. Recognizing how BLAs differ from NDAs allows teams to anticipate challenges early and align their development strategies accordingly.
Getting It Right from the Start
Whether pursuing an NDA or BLA, success depends on clear regulatory strategy, robust clinical evidence, and precise documentation. Regulatory writers play a critical role in ensuring submissions meet FDA expectations—organizing complex data into a format that is both compliant and compelling.
Our experienced team supports sponsors through every stage of NDA and BLA preparation, from gap analysis to full dossier authoring. We integrate seamlessly with your internal teams to keep projects on track and submissions FDA-ready.
Partner with experts who understand the nuances of FDA submissions.
Contact us to discuss how we can help you navigate NDA and BLA requirements with clarity, precision, and efficiency.
In April of this year, FAERS finalized the new process for submitting required safety reports through the Electronic Submissions Gateway (ESG). Read more for how this affects regulatory publishing.
A few months ago, we announced our partnership with Vistatec Life Sciences. This partnership delivers integrated, accurate, translated regulatory writing combined with expert localization expertise that helps organizations accelerate global submissions with clarity, confidence, and compliance.
The European Commission has proposed a series of updates to the EU Medical Device Regulation (MDR) aimed at improving efficiency, reducing burden, and addressing long-standing bottlenecks—particularly around Notified Body capacity, classification clarity, and clinical requirements.
